Every year hundreds of millions of animals are used both by universities and the pharmaceutical industry as experimental subjects. Members of the public who oppose animal experimentation have taken issue with this, pointing out that many experiments are extremely cruel. Scientists have maintained that their experiments are just the price we have to pay if we are going to find new cures for human diseases, leading to an acrimonious debate between the two sides. Now new technological advances have rendered this argument moot.
Animal experimentation is nothing new. It began in ancient Greece, primarily with Aristotle and reached its apogee with the Greek doctor Aelius Galen in the third century CE. When modern biomedical research began to develop in the 17th century it was initially viewed as a hobby carried out by a few wealthy gentlemen. This changed dramatically with the enormous increase in government research funding that followed WWII when science became a real profession. Nowadays millions of scientists perform biomedical research and, as a result, there has been a corresponding increase in the use of experimental animals and in the number of scientific publications where scientists report their results. Unfortunately, it is clear that the vast majority of these publications are rarely cited by anybody apart from their authors and contribute little, if anything, to either scientific discourse or the cure of human diseases.
Since antiquity, scientists have considered animals as stand-ins or models for humans. We don’t do most experiments on humans for ethical reasons; we use animals instead. The idea has always been that if we can cure a disease in an animal then we can cure it in a human. Regrettably, this has rarely proved to be the case. Most diseases have been “cured” in animals many times over, but have failed to impact human treatment, illustrating the fact that for many important purposes animals are poor substitutes for humans and provide inadequate models for human diseases.
Moreover, even when animal research has been useful there have always been questions regarding its ethical appropriateness. In the 17th century, Descartes’ mechanical philosophy encouraged the view that animals were merely unfeeling machines allowing scientists to ignore any potential ethical problems that might arise from their use. Eventually members of the public began to question what was going on. The philosopher Jeremy Bentham (writing in the 19th century) reframed the debate around animal experimentation asking, “The question is not, Can they reason? nor Can they talk? but, Can they suffer?” Since that time, groups who oppose animal experimentation have thought about the issue primarily from Bentham’s point of view. Whatever valuable information they may provide, animal experiments are certainly cruel, and animals certainly suffer.
Now things are changing fast. Since the beginning of the 21st century, new technologies have been developed that enable scientists to use human tissues rather than animal subjects for their experiments, thereby avoiding ethical problems associated with cruelty to animals and producing data that is directly relevant to humans. Gene sequencing studies have allowed scientists to start understanding the molecular fabric of humanity in great detail and have highlighted numerous critical differences between humans and animals. Importantly, the development of stem cell technology has enabled scientists to generate human tissues for an ever-increasing number of experimental purposes. The discovery of human “induced pluripotent stem cells” (iPSCs) has meant that such cells no longer have to be sourced from human embryos, something that was previously viewed as an ethical barrier by many people. The use of stem cells has led to the development of organoids, meaning entire human organs grown from scratch in a laboratory. Even parts of the brain, the most complicated organ in our body, have been grown this way. Organoids from different tissues can be linked together using microfluidic methods producing human-derived tissue arrays of ever-increasing complexity and sophistication. Such human-based organ systems provide much better experimental paradigms for drug development and investigating other biomedical problems than the use of animals. CRISPR-enabled gene editing methods now allow scientists to make genetic changes within stem cells derived from human tissues so that they genuinely reflect the entire spectrum of human diversity, something that also cannot easily be achieved using animals. Additionally, the development of live human imaging methodologies has allowed the assessment of human physiology and pathology in real time. Techniques like these are rapidly making animal experimentation obsolete. Scientists who conduct experiments on animals don’t like to be told that their research is no longer at the cutting edge, but the era of animal experimentation is coming to an end. Animal experiments are looking terribly old fashioned—even to scientists. It’s just not good science anymore.
Over the previous 9 months, everybody’s attention has been completely focused on the COVID-19 pandemic and any progress that might help us put an end to this terrible problem. Now, it seems, that help is on the way in the shape of several vaccines that produce powerful immunity against the SARS-CoV-2 virus. These results represent a triumph for biomedical research. Of course, during the pandemic people have still been suffering from all of the other diseases that plagued humanity before the coronavirus turned up, and research to treat these problems has continued, somewhat in the background, all the while. Nevertheless, exciting things have been going on. For example, over the last few weeks there has been news of new therapeutic advances in the application of gene editing technologies to humans that may herald enormous promise for the future treatment of many debilitating diseases, particularly those that are caused by genetic mutations.
We know, of course, that our genetic makeup determines many aspects of our biology, contributing to things like height and intelligence as well as our propensity for developing different diseases. Such effects can be most easily seen in diseases that are caused by changes in a single gene and which are inherited according to the rules of Mendelian genetics. In these cases, the relationship between the genetic variant and the disease phenotype is very clear. Mutated genes of this type are inherited as recessive or dominant traits. This means that the disease is associated with inheritance of mutant genes from both parents or just one of them. The neurodegenerative disorder known as Huntington’s disease is a good example of a dominantly inherited trait. In this instance, inheritance of the mutated gene from a single parent is invariably enough to produce the disease.
An important disease which is caused through the inheritance of two mutant recessive genes, one from each parent, is Sickle Cell Disease (SCD). SCD results from mutations in the protein hemoglobin which is responsible for the transfer of oxygen around the body in red blood cells (erythrocytes). Interestingly, although these mutations can produce SCD, the trait can also be helpful under some circumstances, which is why it has survived in the human population. It appears that the mutation originated some 7,300 years ago in individuals living under the constant threat of malaria in Sub-Saharan Africa, because it led to some resistance to the effects of the P. falciparum, the organism which is transferred to humans by the bite of a mosquito and is responsible for producing the symptoms of malaria.
The mutation that is responsible for SCD causes the synthesis of what is known as sickle hemoglobin (HbS). HbS is a structural variant of normal adult hemoglobin (HbA). Adult hemoglobin (HbA) is made up of two α- and two β-globin protein chains. HbS is normally the result of a single point mutation (Glu → Val) on the sixth codon of the β-globin gene. Individuals who are heterozygous for the HbS allele (HbAS) carry the sickle cell trait but do not have SCD, whereas individuals who are homozygous for the HbS allele (HbSS) develop SCD, which is characterized by chronic hemolytic anemia, unpredictable episodes of severe pain, and widespread organ damage.
SCD is one of the most common genetic disorders found in the world. An estimated 20–25 million people live with homozygous SCD (HbSS). Approximately 300,000 infants are born annually with SCD. Areas with a high prevalence of malaria such as Sub-Saharan Africa, the Mediterranean basin, Middle East, and India tend to have higher populations of patients affected with SCD. In the United States, approximately 100,000 people—many of them the descendants of the African slave trade—have SCD, accounting for more than 110,000 sickle cell-related hospitalizations annually. There is a wide variability in the clinical severity of SCD, as well as its effects on life expectancy. Although people who carry the HbAS trait (heterozygotes) are clearly resistant to malaria, the mechanism of this effect isn’t entirely clear. Different kinds of biochemical changes have been detected in red blood cells from HbAS individuals, which must make it harder for P. falciparum to establish an infection, but there is no real consensus as to how this occurs. Clearly, SCD is an extremely serious health problem. The fact that it has been maintained in populations such as those in Sub-Saharan Africa, where malaria is endemic, speaks to the fact that just about anything is better than having malaria. In addition to SCD, other individuals carry mutations which make them deficient in synthesizing β -globin chains resulting in an anemia called β-thalassaemia. Each year, 60,000 people are diagnosed worldwide with a severe form of β-thalassaemia, compared with the 300,000 who are diagnosed with SCD.
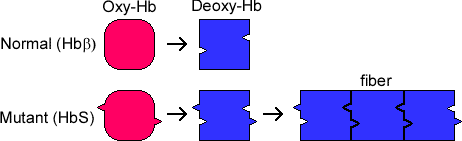
The HbS allele has a lower than normal affinity for oxygen, and the mutant protein has a tendency to aggregate at low oxygen concentrations. Hence, when red blood cells have delivered their oxygen to the tissues and are returning to the lungs via the venous system in an oxygen-depleted state, the hemoglobin of SCD patients polymerizes, which produces sickle-shaped malfunctioning erythrocytes and reticulocytes (erythrocyte precursors).
These cells are sticky and form clumps that occlude blood vessels, particularly small and some large vessels producing what is known as a vaso-occlusive crisis (VOC) which causes ischemic injuries. The sickled red blood cells are also brittle and burst, producing profound hemolytic anemia. During a VOC, the most common complaint is pain, and recurrent episodes may cause irreversible organ damage. One of the most severe forms is the acute chest syndrome which occurs as a result of infarction of the lung parenchyma. This can rapidly result in death.
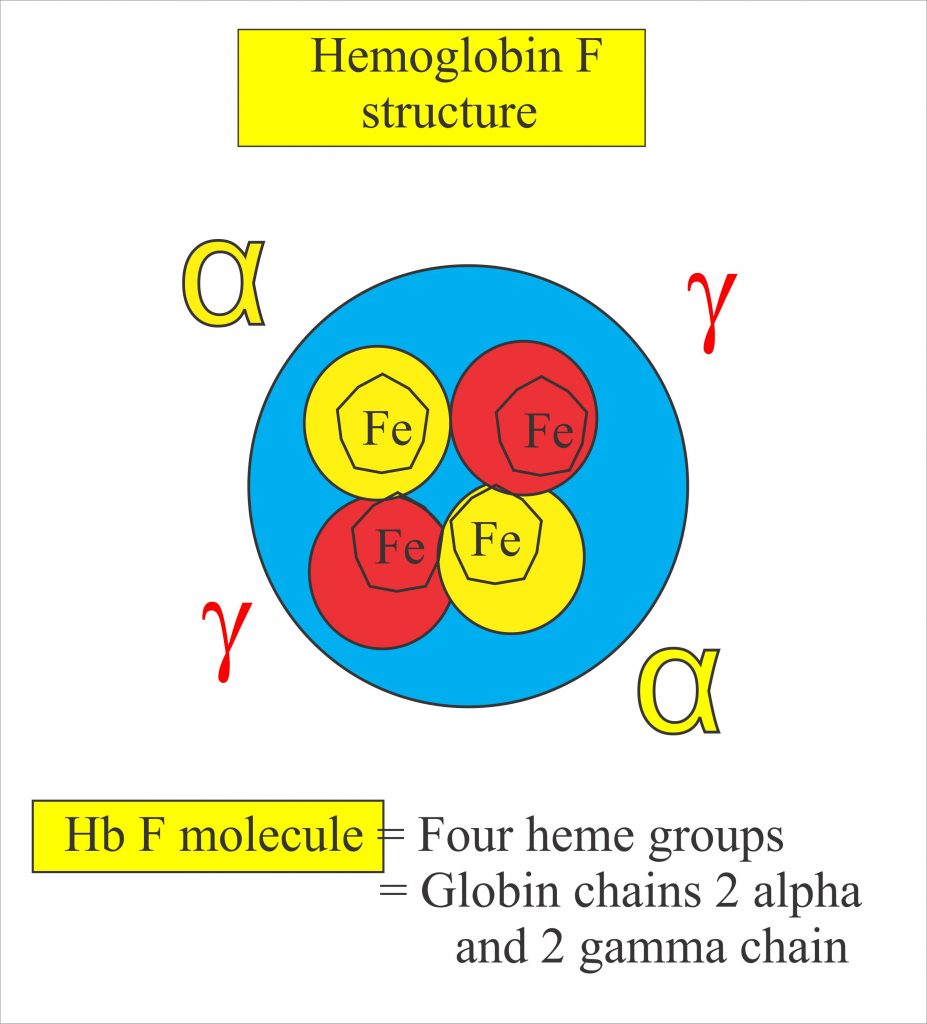
For a patient with SCD, both chronic pain and intense bouts of acute pain are particularly hard to deal with, and a great deal of pharmacological research has attempted to find ways of dealing with the pain that does not depend on the use of opioids. Nerves that sense pain innervate blood vessels and can be activated by inflammatory mediators and other processes occurring during a VOC. Drugs that interfere with these processes may be helpful in treating pain occurring in SCD patients in particular. Recently, there has been progress in achieving this aim. The first new therapy to be introduced for treating SCD was hydroxyurea (or hydroxycarbamide), which is the only drug frequently employed in SCD management that is approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Hydroxyurea’s principal mechanism of action is stimulating the production of HbF. HbF is a fetal form of hemoglobin which is not mutated in the disease and is the major form of hemoglobin that humans produce up until around 6 weeks of age. HbF is associated with the synthesis of γ-hemoglobin, which reduces HbS polymerization and so ameliorates pathological processes such as sickling, VOCs, and events downstream of these triggers, including pain.
The mechanism by which hydroxyurea induces HbF generation is unclear, but by increasing fetal hemoglobin concentrations in erythrocytes and reticulocytes, hydroxyurea treatment has significantly reduced the rates of VOCs, subsequent hospitalizations, and mortality in a range of patients. Unfortunately, despite these benefits, adherence to a hydroxyurea regimen has been problematic for some patients. Adverse effects of the drug include bone marrow suppression, large inter-patient variability in the effects it produces, and variations in the maximum tolerable dose. Consequently, other therapies have been sought based on a detailed understanding of the molecular and cellular events that produce VOCs. Other treatments for SCD include an antibody against the molecule P-selectin that is involved in the aggregation of cells in blood vessels that produce VOCs, or even complete replacement of the stem cells (which generate erythrocytes) in the patients’ blood with immunologically compatible donor stem cells that do not carry mutated hemoglobin. While these treatments have been helpful for a large number of patients, they do not fix the basic problem: the expression of mutant hemoglobin that produces the disease. However, the effectiveness of hydroxyurea which results from its ability to induce HbF is a clue to the success of the new genetic therapies that have been announced over the last few weeks.
A true cure for SCD would involve altering human genetics in some way so that HbS was no longer produced. Until recently, one could only dream of actually changing the structure of a gene in a live patient—something that seemed more like science fiction than therapeutic medicine. However, several new technologies have now been developed which actually make such treatments a real possibility. The most famous of these techniques relies on what is known as CRISPR/Cas9-mediated gene editing, a technique that has been very widely described in the popular press and which garnered the Nobel Prize for the two lead scientists responsible for its discovery. CRISPR/Cas9 is a molecular complex that allows an enzyme named Cas9 to cut the DNA of genes at specifically targeted points. Potentially, this might allow the gene to be inactivated or changed (edited) depending on exactly how the technique is employed. CRISPR/Cas9 was actually discovered in bacteria where it is used to defend cells against attack by viruses, but CRISPR/Cas9 methodology has now gone through various modifications that allow it to be used in human cells.
The idea of using CRISPR/Cas9 to make therapeutic changes to the human genome is a fantasy that has proved to be almost irresistible for many scientists, although at this time, there is supposed to be a moratorium on doing this until all aspects of the methodology have been shown to be safe. Nevertheless, its use lead to a huge scandal in 2018 when a Chinese investigator name He Jiankui reported that he had created CRISPR/Cas9-mediated gene-edited embryos for two twin girls. He intended to inactivate a gene called CCR5, which is involved in enabling HIV-1 pathogenesis, in order to prevent the twins being infected by the virus which had infected their father. The unanticipated announcement of this procedure created an enormous furor and many scientists, bioethicists and others took He Jiankui to task for performing his experiments. In the end, He Jiankui lost his job, was fined and given a 3 year prison sentence. And really, he was jumping the gun because the accuracy of CRISPR/Cas9 editing in this context had not been fully investigated, and subsequent studies demonstrated that it can produce a large number of unintended “off target” edits, which might be highly problematic, to say the least. One scientist said of these new revelations, “If human embryo editing for reproductive purposes, or germline editing, were spaceflight, the new data are the equivalent of having the rocket explode at the launch pad before take-off.” Investigators are now likely to be much more circumspect before trying things like this on human embryos. But that was two years ago. Now, further investigations using CRISPR/Cas9 appear to show that it can indeed be used effectively and safely in the treatment of SCD in particular. Recently, two papers appeared in the New England Journal of Medicine (NEJM) which both use “genetic engineering” techniques to intervene in SCD. One of these used CRISPR/Cas9 and the other used another powerful technique known as RNA interference (RNAi), another Nobel Prize-winning phenomenon that was originally discovered using the nematode worm C. Elegans and has subsequently been adapted for human use.
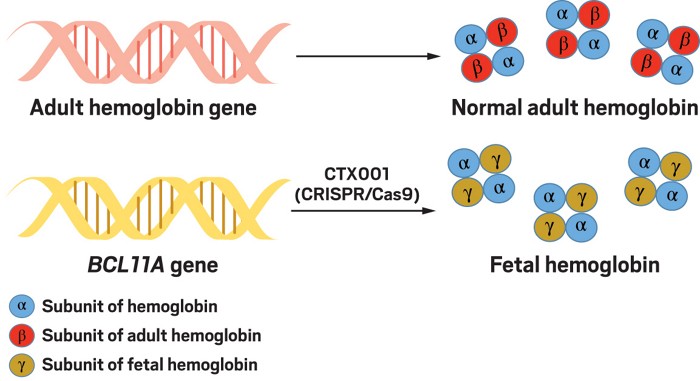
As we have seen, the drug hydroxyurea is helpful for treating SCD because it allows adults to synthesize HbF. Because this gene does not carry the SCD mutation, HbF can act as a replacement for mutated HbS. In the fetus and newborns, HbA protein is produced at very low levels because the erythrocytes have not yet shifted from expression of the HbF gene to expression of the HbA gene, a phenomenon known as “hemoglobin switching.” The two new therapies asked a simple question: what would happen if one could use a genetic trick to allow HbF to be activated in SCD victims? One interesting observation is that individuals who develop SCD show no symptoms when they are young and HbF is still being expressed, that is prior to it being turned off and the synthesis of HbS being turned on instead. Furthermore, in rare individuals, HbF continues to be made in adulthood and these individuals display resistance to the effects of coinherited HbS—they are free from all of the symptoms of SCD. All of these results, together with the known effects of hydroxyurea, strongly suggest that strategies that serve to maintain higher levels of HbF in adulthood should be effective in countering the effects of HbS. But how could this be achieved? Why not try to disable the switch that is responsible for turning HbF off and HbS on?
One key question therefore is what is the switch made of? A clue as to its identity came from genome wide association studies (GWAS) which identified single-nucleotide polymorphisms (SNPs), small genetic alterations, associated with increased expression of HbF in some adults. Many of these SNPs were found to be located in a gene on chromosome 2 that encodes a protein called BCL11A. These SNPs were also found to be associated with a lower severity of β-thalassaemia and SCD. BCL11A is what is known as a zinc finger-containing transcription factor, a protein that normally represses HbF, γ-globin expression and fetal hemoglobin in erythrocytes. The SNPs that are associated with fetal hemoglobin production are located in a particular part of the gene called an erythroid-specific enhancer, which allows BCL11A to be made. These SNPs interfere with the ability of the enhancer to work efficiently and result in downregulation of BCL11A expression and consequently increased expression of fetal hemoglobin.
Based on these findings, one of the new papers reported attempts to use CRISPR/Cas9 to inactivate BCL11A in one SCD patient and one patient with β-thalassaemia, both of whom should theoretically be helped if they increased their activation of HbF.
The clinical trial proceeded as follows. First of all, the authors obtained stem cells from the blood of these patients which could be identified and purified because they express a protein called CD34. They then expressed the CRISPR/Cas9 enzymatic complex in these cells, which was guided to the BCL11A enhancer sequence through a specially designed guide RNA molecule—this is consistent with the normal method by which CRISPR/Cas9 functions. When they examined the resulting cells, the authors observed efficient editing of the desired region and—importantly, given the above discussion—little evidence for off-target modifications of other genes. In other words, it seemed as though the gene editing that they had produced was extremely specific. The patients were then given a treatment to wipe out any remaining mutant stem cells that were still present in their blood/bone marrow which were then replaced by infusion of the new CRISPR/Cas9 modified stem cells. After a few weeks, there was clear evidence that these new stem cells had successfully engrafted themselves into the bone marrow of the recipients. Importantly, as hoped, both patients exhibited long term increases in their levels of HbF protein. What is more, their diseases were clearly in remission. More than a year later, both patients had high levels of modified stem cells and increases in fetal hemoglobin. The patient with β-thalassaemia did not need a single blood transfusion during that time, and the patient with SCD was completely free of VOCs. In other words, the treatment worked just as planned. Although these results only involved two patients, apparently further patients have now been treated to good effect.
The second paper published in NEJM achieved similar results by using a somewhat different technique. Rather than turning off the switch that normally blocks the synthesis of HbF, the authors devised a strategy for inhibiting the synthesis of the BCL11A protein. Fire and Mello won the Nobel Prize in 2006 for discovering the phenomenon of RNA interference (RNAi). This phenomenon involves the synthesis of small hairpin-shaped RNA (shRNA) sequences that bind to messenger RNAs (mRNA), and initiate their destruction, thereby downregulating the synthesis of the protein that the mRNA normally directs. The process is very important in normal biology but has been subsequently adapted as a useful tool that can be employed for the targeted downregulation of any particular mRNA, resulting in a reduction in levels of specific proteins. In this case, the idea was to inhibit the synthesis of the BCL11A protein using this method. In order to achieve this aim, stem cells were isolated from the patients and were infected with a lentiviral vector which had had the appropriate shRNA that targeted BCL11A mRNA inserted into its genome. In this case, the inactive lentivirus acts like a ballistic missile enabling the shRNA sequence to be inserted into the stem cell genome. Six months later, all six of the treated patients exhibited high levels of HbF and a complete absence of VOCs. In other words, although the two studies had disabled BCL11A function using two different methods, both methods had resulted in long-term elevations in HbF and seemingly complete remission of SCD (or β-thalassaemia in one patient). Moreover, this occurred in a seemingly highly specific manner, free of the effects of non-specific off-site editing that might have been expected. One thing to note is that these experiments were not designed to inactivate BCL11A in the entire body—only in erythrocytes—and so differ from the experiments performed by He Jiankui which, because they were performed on a fertilized egg, essentially knocked out the targeted CCR5 gene in every tissue.
These results are extremely dramatic, exciting and imply that gene editing may be widely used in the future to great effect in the treatment of numerous diseases. For example, one of the companies that was involved in these studies has now targeted Glycogen storage disease type Ia, Duchenne muscular dystrophy, myotonic dystrophy type 1 and cystic fibrosis for CRISPR/Cas9-mediated therapies.
Since the dawn of man, therapies for diseases have usually employed different types of drugs. These new experiments herald something entirely different—changing the actual fabric of humanity by altering the genome itself. This is not only a fantastic medical advance but, in some respects, is also the stuff of science fiction. Nevertheless, there is every reason to expect genetic approaches such as these to become more and more common in the not too distant future.
References
Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, Daley H, MacKinnon B, Morris E, Federico A, Abriss D, Boardman K, Khelladi R, Shaw K, Negre H, Negre O, Nikiforow S, Ritz J, Pai SY, London WB, Dansereau C, Heeney MM, Armant M, Manis JP, Williams DA. N Engl J Med. 2020 Dec 5. doi: 10.1056/NEJMoa2029392.
CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, Ho TW, Kattamis A, Kernytsky A, Lekstrom-Himes J, Li AM, Locatelli F, Mapara MY, de Montalembert M, Rondelli D, Sharma A, Sheth S, Soni S, Steinberg MH, Wall D, Yen A, Corbacioglu S. N Engl J Med. 2020 Dec 5. doi: 10.1056/NEJMoa2031054.