
Like many people, I often enjoy watching a good TV series in the evening. I really like crime shows and, of course, there are plenty of excellent ones. A case in point is the British TV series “Unforgotten.” Once I started to watch it, I was glued to the TV for the entire 3 series run. Each series revolves around the discovery of buried bones—for example under a freeway, in a river or in the basement of a house. But whose bones? As it turns out, they all belong to people who went missing several decades ago and nobody ever found out why. In other words, these are cases that have been forgotten. But Detective Chief Inspector Cassie Stuart and her sidekick, Detective Inspector Sunny Khan, want to set things right and figure out who these people were, what awful things might have happened to them and who was responsible. They want the cases to be “unforgotten” and the perpetrators of the crimes brought to justice.
As it turns out, the theme of forgetfulness not only concerns Cassie’s role as a detective but also her personal life. Cassie lives with her father who is getting on in years. Now he has started to forget things. But is this just normal forgetting or is something more serious involved? Perhaps this is the start of senile dementia? Cassie and her Dad argue about whether he should be tested or not. The Dad says he is fine—Cassie isn’t so sure. And, of course, this is a scene that is played out in many families all over the world. Dementia is a terrible fate, a long decline into a cognitive void where eventually everything is forgotten. But is “unforgetting” for these dementia victims a medical possibility? The recent announcement of the first treatment that might actually slow the cognitive decline in patients with Alzheimer’s disease has been hailed as a major breakthrough and has generated great excitement in the media.
It has been argued that the group of syndromes termed “senile dementias” are the most serious health problem afflicting the entire planet. Some 50 million people throughout the world suffer from dementias which affect the victim’s ability to remember things and to function effectively in everyday life. Paranoia and personality changes also often occur. Alzheimer’s disease (AD) is by far the most common form of dementia, making up some 70% of the victims. Other common dementias include Lewy body dementia, vascular dementia and frontotemporal dementia. Overall, the cost of treating AD throughout the world has been estimated at somewhere around a billion dollars (to be precise, in 2015 it was actually calculated as being $818 million). Unfortunately, although we now have a much better idea as to the natural history of the disease and its potential mechanisms, no disease-modifying interventions are available, only some medications that produce a small improvement in symptoms. This isn’t from want of trying. Until recently, most “Big Pharma” drug companies had very active discovery programs in this area. But with no effective new drugs on the horizon, they are beginning to lose heart and many of these programs have been discontinued. Of course, it’s a difficult problem. The disease is very long lasting, so it is hard to assess whether a potential therapy is really working or not without running sophisticated clinical trials over long periods of time—and, of course, these are extremely expensive. Hence, the announcement that something might actually be working is reason for considerable excitement for patients and researchers alike.
To understand what is going on, we need to take a look back at exactly what we know about the causes of AD. Naturally, there are reports of people becoming demented throughout history, particularly elderly people. However, the study of dementias as a part of modern medicine began on 25 November, 1901, when a 51-year-old woman named “Auguste D.” (actual name, Auguste Deter) was admitted to the Municipal Asylum for Lunatics and Epileptics in Frankfurt, Germany. The doctor who examined her was Alois Alzheimer. Among the things that Alzheimer noted was that the patient appeared extremely paranoid and deluded, imagining that her husband was having an affair with a neighbor. She also had difficulty remembering things and performing everyday tasks like cooking. Her condition worsened over time, and eventually, she was even unable to recall things that had only recently occurred. Auguste D. remained in the institution until her death in 1904.

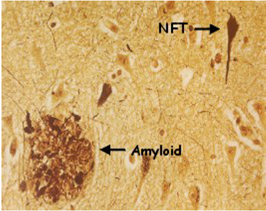
Alzheimer, an expert neuropathologist, subsequently examined Auguste D.’s brain and described some striking abnormalities. These included the presence of the “neurofibrillary tangles” (NFT), “amyloid plaques,” and nerve degeneration which are now known to constitute the cardinal pathological hallmarks of the disease. Further patients suffering from similar symptoms with the same brain pathology were then recognized, and in 1910 the disease became officially known as Alzheimer’s disease. Originally, AD was defined as a disease in which young people inappropriately developed the cognitive problems associated with old age. However, it was eventually realized that the disease defined by its similar cognitive and pathological signs actually affected both young and old people alike. By the 1960s, it was becoming apparent that the brains of all patients with AD exhibited tangles and plaques. Interestingly, however, there were also examples of elderly people who possessed these pathological signs but were not demented, making the precise relationship between the observed neuropathology and dementia unclear. On the whole, however, the increased occurrence of plaques and tangles seemed to correlate with the seriousness of dementia. But were the protein constituents of tangles and plaques actually responsible for producing AD or were they just features otherwise associated with the course of the disease?
Clues as to what was going on came from the study of human genetics. It had been recognized as early as the 1930s that there were some families in which several people developed AD early in life with a pattern suggesting a Mendelian autosomal dominant form of inheritance. It was also observed that patients with Down’s syndrome, who have an extra copy of chromosome 21, always seemed to develop AD at some point in their lives. Around this time, a protein called the amyloid β-protein 1-42 (Aβ) was identified as being the major protein component that made up the core of the amyloid plaques found in AD brains. This small protein was found to be derived from the cleavage of a larger transmembrane protein, subsequently named the amyloid precursor protein (APP) which, interestingly enough, was found to localize to chromosome 21. Mutations in this gene were also linked to some cases of early onset AD. In other cases, mutations in the presenilin gene, located on chromosome 14, were linked to early onset AD. Presenilin was found to be part of an enzyme complex eventually named γ-secretase that was responsible for making one of the cuts that releases Aβ from APP. The second cut that released Aβ was found to be made by another enzyme named β-secretase. It is thought that this pattern of APP cleavage is ultimately pathogenic. Under other conditions, APP can be cleaved by an enzyme called α-secretase that precludes it being cut by β-secretase; this cleavage pattern does not lead to the generation of Aβ and does not contribute to the production of plaques. It is now thought that monomeric Aβ 1-42 is soluble. But the protein has a marked tendency to aggregate and easily forms insoluble aggregates called oligomers. One popular hypothesis is that these oligomers are the molecular species responsible for producing the pathological changes in the brain that are typical of AD.
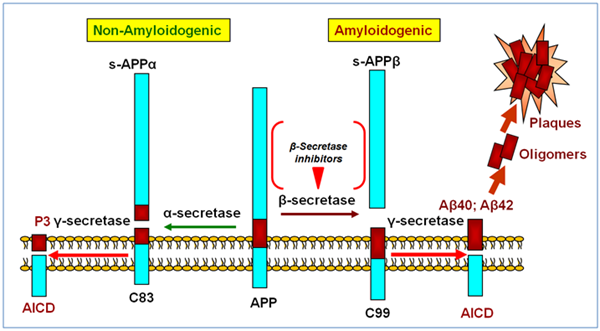
As far as the tangles are concerned, they were found to be composed of the microtubule-associated protein tau, which is normally expressed in nerve axons but in AD becomes highly phosphorylated and aggregated into abnormal parahelical filaments (PHFs) in neuronal cell bodies. Over the years, there has been a lively debate as to whether Aβ or tau is responsible for the brain changes seen in AD. The group favoring Aβ became known as “Baptists” and the group favoring tau became known as “Tauists.” Although it is still not entirely clear which group is correct, most potential therapies aimed at treating AD have concentrated on the Aβ hypothesis which imagines that Aβ is the main culprit.
But if that is the case, what kind of therapy might be effective? The first idea to be floated was that drugs which blocked either the β- or γ-secretases that were involved in the release of Aβ from its precursor protein in the first place would do the job. A lot of time and effort was put into developing molecules that would perform this task but unfortunately, although they seemed to inhibit their target enzymes effectively and reduce the amount of Aβ in the brains of experimental mice or humans, clinical trials showed that they weren’t effective in treating the disease. Of course, it should be realized that clinical trials to test drugs for treating AD are themselves actually rather complicated. Do we expect a 90 year old with advanced dementia to actually get better? How about a younger person who is just starting to exhibit symptoms? What does “better” mean anyway? That the patient becomes normal again? Or perhaps that the rate of cognitive decline is slower? Exactly when to test a therapy for AD and what the expectations are for the drug involved is clearly an important consideration. If a potentially useful drug was tested on a group of patients where the cause of the disease had already produced irreversible damage, then no benefit would be observed whereas effects might have been seen if the drug had been tested earlier on in the course of the disease. Overall, considering the fact there are no therapies whatsoever for AD, most people think that a significant slowing of the rate of cognitive decline would be considered an important first step.
Advances in the science of human genetics have started to shed further light on the potential causes of AD and so suggest further potential routes for therapeutic intervention. After all, most patients who suffer from the disease don’t have the extremely aggressive early onset form of AD that is associated with “dominant” mutations in APP or presenilin that are inherited in a clearly Mendelian fashion. Rather, the major group of AD patients may have versions of many different genes that give them a greater “susceptibility” for getting the disease. In fact, for most people, whether they get AD or not ,is probably the result of a combination of these relatively modest genetic effects. Hence, knowing which genes produce these more subtle effects is clearly very important in the grand scheme of things. For example, quite some time ago it was shown that people who had a particular version of the apolipoprotein E gene were more likely to get AD and now there are a considerable number of such “risk” genes that have been identified. Each one might tell us something interesting about the natural history of the disease. Consider the gene that codes for the protein TREM2. TREM2 is usually expressed by cells called tissue macrophages that are central components of what is known as the innate immune response. Innate immunity is the body’s first response to anything that disturbs tissue homeostasis. This could be a toxin, an infectious agent, stress proteins expressed as warning signals by cells, or the detritus of dying cells. In the brain, tissue macrophages are called microglia. One thing that microglia can do is act as cellular garbage dumps that mop up pieces of dying cells or inappropriately aggregated proteins such as Aβ. It seems as though the protein product of the TREM2 gene has an important function in keeping microglia healthy so that they can go about their business of clearing up the brain. Mutations in TREM2 that are associated with AD risk seem to reduce the ability of TREM2 to do its job. Now, consider, if one of the things that microglia do is to remove aggregates of Aβ in the brain, then in people with TREM2 mutations, levels of Aβ would build up in the brain. In other words, accumulations of Aβ might be due to increased production of the protein through changes in the activity of the β- and γ-secretases, or to reduced ability to remove the protein as a result of something like a TREM2 mutation, or perhaps both things.
If accumulation of Aβ in the brain is an important factor in causing the symptoms of AD, then how exactly does this happen? Here again there are several ideas. One is that the formation of amyloid plaques is somehow toxic. After all, they are observed in flagrante delicto surrounded by the extensions (axons and dendrites) of dying nerve cells. On the other hand, it has also been argued that the plaques are really just garbage dumps that collect Aβ and that it is small aggregates of Aβ known as “oligomers” that actually produce the toxicity. At any rate, all of this suggests that rather than blocking the production of Aβ, if you could find a way of mopping it up and getting rid of it then this might also be a valuable therapeutic approach. How about using an antibody? Antibodies are usually used by the immune system to target problematic molecules associated with infectious agents or other foreign objects. They can do this with a great degree of specificity. An antibody that recognized Aβ might be able to remove it from the brain and get rid of it by a number of routes the body has at its disposal for destroying antibody/antigen complexes. This approach has been considered very seriously as a potential treatment for AD. If the antibody were given just as the disease was beginning to appear (known as the prodromal phase), it might prevent the entire cascade of resulting toxic events. We know from brain imaging studies that amyloid plaques begin to appear in the brain well before cognitive symptoms are seen in affected individuals. So, the theory is that intervening at this early stage might prevent the entire cascade of Aβ-related events from occurring—or, at least, intervening at an early stage of symptomatic disease might prevent its progression.
Here again there are a number of ways one could approach this problem. One type of immunization is known as “active” immunization: an appropriate antigen, a form of Aβ in this case, would be injected into a patient and over time the recipient’s immune system would generate antibodies against Aβ. This may take a while but might afford lifelong protection or at least very long-lasting immune protection against the effects of Aβ. Another kind of immunization is known as “passive” immunization. In this case, active antibodies are generated in a laboratory or other facility and then injected into the recipient. Such antibodies will begin working immediately but protection will wane as they are used up and then a further injection will be necessary. Many potent therapies these days rely on this method. For example, passive immunization against the cytokine TNF-α in preparations like Remicade are widely used for the treatment of inflammatory diseases. Overall, “biological” reagents like this have really revolutionized pharmacology in recent years.
Both active and passive techniques have been investigated in the treatment of AD. Generally speaking, the results have been disappointing. But one has to be careful here. Exactly which entity do you want your antibody to target? Given what we know about the natural history of plaque formation in AD, we would probably want to have an antibody that targeted aggregated forms of Aβ found in oligomers or plaques, rather than the soluble monomeric form of the protein. With this possibility in mind, a few years ago a company called Neuroimmune set out to find such an antibody by examining the white blood cells from older people that were not afflicted by AD. Maybe they already had such antibodies? The company, in collaboration with a second company called Biogen, obtained a positive result and prepared the antibody, which they named aducanumab, so that it could be used in clinical trials.
Biogen actually sponsored seven clinical trials investigating aducanumab in humans. The interim results from the first 165 patients participating in this trial were published in the high profile scientific journal Nature. All doses of aducanumab (given as monthly infusions into the bloodstream) were seen to significantly reduce amyloid plaques in the brain in a time- and dose-dependent manner compared with before treatment, whereas little to no change was seen in the placebo group after one year. The greatest reduction in plaques was seen at the higher doses. Aducanumab also appeared to slow the rate of cognitive decline. So overall, these Phase 1 trials were very promising and Biogen continued testing, continuing with Phase 2 and 3 trials—the ultimate test of a drug’s efficacy in the target human population. The phase 3 trials were aimed at assessing the efficacy of aducanumab, given once a month at low and high doses by infusion into the bloodstream, in treating the symptoms of Alzheimer’s disease. Based on the interim data, the hopes were high. However, an independent data-monitoring committee found that, based on the initial data coming out of the trial, the outcomes were unlikely to meet their primary objective, and so in March 2019 the trials with aducanumab were halted. That seemed to be the end of things for aducanumab. Or was it?
As it turned out, the decision to halt the trials hadn’t considered some of the data that were obtained later on in the trials. These data seemed to show that patients who had continued to take the drug at its highest dose did show a fairly impressive slowing in their rate of cognitive decline (23%). This was actually very promising and just a few weeks ago, the company made a rapid volte face declaring that they would, after all, be presenting their results to the Food and Drug Administration with a view toward bringing it to market. Although we don’t know if the FDA will approve the drug or how successful it will be, this is the cause of all the excitement as it would be the first example of a disease-modifying drug for the treatment of AD. It should be mentioned that patients who are treated with aducanumab can suffer from side effects including a drug-induced syndrome called ARIA (amyloid-related imaging abnormalities), headaches, urinary tract infections, and upper respiratory tract infections. However, if aducanumab works as advertised, it might be just the right option for somebody like Cassie Stuart’s Dad who is just starting to exhibit signs of what could be AD. In other words, there may be a lot of time during which an intervention like aducanumab could be used to significantly reduce the rate at which the disease takes hold.

For more serious victims of AD, however, the picture is still bleak and may always be that way. Nevertheless, some things may help. If you can’t use aducanumab you can at least get a cat. It has been widely reported that companion animals have really significant calming effects on demented elderly patients. Furthermore, cats are much cheaper than a course of aducanumab treatment and are unlikely to give you ARIA or other side effects. Give me the cat!