
News concerning the “Opioid Crisis” has been extremely widespread over the last year and nearly everybody these days will be at least somewhat familiar with what is going on. A combination of factors, including over-prescription of opioids under inappropriate circumstances, have helped to fuel the crisis which is now responsible for the deaths of more than 70,000 people in the USA every year, even as we come to understand the phenomenon better and engage in better ways to stop it. There are numerous aspects of the crisis that demand our attention, but one which is of paramount importance concerns the properties of opioid drugs themselves. Morphine and other opioids are the pain killing drugs par excellence and they are indispensable to many areas of medicine such as surgery and anaesthesia. However, in addition, we know that outside a strict clinical setting the drugs are extremely dangerous. They are highly addictive and are easily abused. They also have powerful effects on respiration. Opioid inhibition of breathing is the reason why people actually die from taking high doses of these drugs. Because opioids generally work so well as pain killers, there has been a tendency to hand them out in a “one size fits all” fashion for treating all kinds of pain. This is the case even though their chronic use is dangerous and, in fact, they don’t work all that well for treating some chronic pain syndromes. So, the question arises –Why don’t we just make better opioid drugs? Why don’t we make drugs that kill pain as effectively as opioids but are free from their addictive and other dangerous side effects? Come on scientists, how hard can it be to do that?
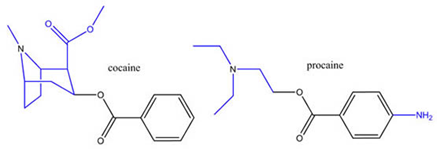
The idea that this was an achievable goal arose in the 19th century, fueled by results on the drug cocaine. Cocaine is an addictive psychostimulant drug but it is also a good local anaesthetic. So, why not separate these two activities? Note that the chemical structure of cocaine consists of a “tropane” group attached to a “benzoic acid” group. What would happen if the two were separated? When this was done by scientists at the Hoechst drug company, they found that derivatives of the benzoic acid portion alone, things like procaine and lidocaine, worked very well as anaesthetics but were free of psychostimulant activity. Job done! So, all we needed to do was the same kind of thing with morphine and obtain a really good analgesic drug that was no longer addictive or suppressed breathing. Unfortunately, this aim has proved to be much harder to achieve. In fact, nobody has ever been successful in achieving it—and thereby hangs a tale.
Attempts to improve the profile of drugs like morphine began in the 19th century once the first pharmaceutical companies had been established. Researchers at these companies began to use the tools of the newly developing science of organic chemistry to modify the structure of morphine. In 1897, researchers at the Bayer company in Germany, who had had good luck acetylating salicylic acid to produce aspirin, tried the same thing on morphine and produced diacetyl-morphine. On performing some self-experimentation, one of the scientists declared that the drug made him feel very strong and powerful, or “Heroisch” in German. The name stuck and the drug became known as heroin. Heroin was originally marketed as a cough suppressant, like morphine and codeine, but, in contrast to these drugs, it was supposed to be non-habit forming. This is obviously quite untrue and one wonders how they ever came to such a conclusion. The reason, it appears, was that the drug was mostly used to treat tuberculosis patients. As their condition never improved, patients never really stopped taking heroin and so their dependency on the drug wasn’t really evident. Nevertheless, after a few years, the first heroin addicts started to appear and the real dangers of the drug became manifest.

However, this original rather spectacular failure has never dissuaded people from trying to produce non-addictive but effective opioid analgesics, even prior to the present crisis. The obvious attraction of such a drug to the pharmaceutical industry and humanity in general will be clear. Over the years, hundreds of papers have appeared in the scientific literature claiming to have solved the problem. These papers have often received a lot of attention and have been published in important “high profile” scientific journals, which garnered the authors a huge amount of science recognition. Inevitably, however, they all ultimately ended up in disappointment when tested further.
Let us examine a few of these attempts to try and understand what is going on. First of all, a few words about how opioid drugs work. All of these drugs activate a receptor called the μ-opioid receptor (MOP), which is expressed by nerve cells at various points up and down the neuraxis, including nerves that are situated at various points in the spinal cord and the brain, as well as nerves in the peripheral nervous system that are involved in sensing pain. MOP is one of a family of 4 receptors that are responsible for mediating the actions of the peptide neurotransmitters known as endorphins or related neuropeptides known as nociceptins. Apart from MOPs, there are also δ (DOP) and κ (KOP) opioid receptors and a nociceptin receptor (NOP). When these receptors are activated by agonists, they produce biochemical signals that reduce the firing rate of neurons and inhibit neurotransmitter release. It just so happens that, out of all the hundreds of receptors that exist in the genome, MOPs have a unique expression pattern that allows them to produce the profound analgesic effects that are observed. So, as far as we know, activation of no other receptor will be able to produce the same array of effects on the nervous system that would allow a drug to be as effective an analgesic as something like morphine. This doesn’t mean that drugs that activate other receptors, such as receptors for cannabis, might not produce some useful analgesic effects, but they are unlikely to match the profound analgesic effects observed with opioids that have resulted in these drugs becoming the cornerstone of surgery and pain treatment in general. Hence, trying to make a better version of morphine has a lot of appeal.
Attempts to achieve this aim fall into several categories. The first category might be called “Let’s see what Nature has come up with.” After all, Nature came up with morphine and codeine which are derived from poppies, and so maybe it also came up with other things as well. Perhaps, we just need to look around carefully. Moreover, human beings have had tens of thousands of years, at the very least, to experiment with plants and other natural products, so if there was something out there we might well have stumbled across it. In fact there are such things. The best example is the tree Mitragyna speciosa, a tropical evergreen tree in the coffee family native to Southeast Asia. Extracts of the leaves of this tree, commonly known as kratom, have been used as a traditional folk medicine in Southeast Asia. And kratom really does seem to be effective. The tree produces chemicals, particularly mitragynine and 7-OH mitragynine, which, when tested, appear to be perfectly good MOP agonists and produce effects that are strikingly similar to those of morphine, although, interestingly, their chemical structures are quite different from other known classes of opioids. Mitragynine and 7-OH-Mitragynine can produce strong opioid-like analgesia, but they are also addictive and produce respiratory depression just like other opioids. In spite of the fact that kratom is very similar to morphine, its use is more or less legal in most parts of the USA (depending on the state) and deaths due to respiratory depression appear to be rare. It should be recognized that kratom is usually used as a tea or other type of extract and so it is unlikely that, in everyday use, it ever reaches the doses that would produce really dangerous morphine- or heroin-like effects, and so some benefit may be obtained by using kratom tea for general pain relief. However, it is unlikely that kratom is actually a “better” drug than morphine in terms of its core pharmacological properties. It would also be wrong for people to think that kratom is some “natural” non-dangerous alternative to morphine. Really, it’s just another opioid as far as anybody can tell and so it is as potentially dangerous as any other opioid.

Another kind of approach to making a superior version of morphine involves trying to get morphine to work better by playing around with its chemical structure and resulting pharmacological properties, as was attempted during the original synthesis of heroin. There are several types of theories in this area. One hypothesis goes this way: When MOPs are activated, they produce different kinds of biochemical signals in nerve cells. One kind of signal is called G-protein activation. Another kind of signal is called β-arrestin activation. Perhaps, it is argued, one kind of signaling is responsible for the good effects of opioid drugs, such as analgesia, and another kind of signaling is responsible for the bad effects, like addiction and suppression of breathing. Drugs like morphine produce both types of signaling and therefore produce both good and bad effects. However, our experience with receptors like MOPs is that it should be possible to synthesize molecules that only activate one signaling pathway or the other. These types of molecules are called “biased” agonists. So, what would happen if we made biased agonists for MOPs: would the good/bad idea really be true in practice? The results are that it is. Or, at least, that was how the story was originally told. Several publications reported results showing that mice in which the gene for β-arrestin had been “knocked out”, still exhibited strong morphine-induced analgesia, but did not exhibit effects like morphine-induced respiratory depression. This suggested that MOP agonists that were “biased” towards producing G-protein but not β-arrestin activation should be free of dangerous side effects. Buoyed by these exciting results, drug researchers launched an intense search to find such molecules and were successful in reaching this goal. Several reports demonstrated that MOP agonists that were biased towards G-protein activation did, indeed, produce considerably less in the way of respiratory depression, constipation and other side effects but were still good analgesics. So, it seemed that the great search for an improved opioid drug had at last been successfully concluded. Unfortunately, when the leading drug in this category (TRV-130, or Oliceridine) was tested in humans in a clinical trial, the promising results didn’t seem to hold up and the drug appeared to be just another MOR agonist. So, what happened? The problem here, which is certainly not unique to this set of investigations, but has often plagued the search for improved MOP agonists, is what is called lack of “translation.” This can mean a couple of things. In the first case, it means that results found in one laboratory do not “translate” to another laboratory. In other words, an initially exciting result fails to be repeated in other laboratories and eventually fizzles out. Why does this happen? Nobody knows, but it probably has more to do with the sociology and culture of science than science per se. Another type of lack of translation refers to the inability of results obtained in rats and mice to translate to humans. This is another common observation that is certainly not unique to the opioid field. The fact is that pain is a complicated phenomenon. There are triggers for pain that elicit rapid reflexive movements of limbs away from potentially tissue damaging events like the intense heat of a flame, strong acid or sharp objects. Secondly, there are affective, conscious aspects of pain that are enabled by higher conscious brain function. As mentioned above, MOPs are uniquely distributed in the nervous system so that they dampen all aspects of pain, both reflexive and affective, and so they are extremely effective overall. But what is pain to a mouse? This question echoes Thomas Nagel’s famous 1974 essay “What is it like to be a bat?” We don’t really know the answer to this question. A mouse has an inner life and presumably feels something akin to pain, but its appreciation of this phenomenon might not be the same as that of a human being. Hence, its reactions to drugs that involve effects on the higher regions of the central nervous system responsible for consciousness and cognition may well differ from those of a human. Indeed, it is a general problem in the field of drug research that mice are not good models for human cognitive functions and that drugs that show effects in mice seldom translate exactly to humans in these kinds of situations. Tests in monkeys may be more reliable, even though monkeys aren’t exactly human either.
Another approach to the problem is to produce drugs that can modify the effects of MOP activation through a separate pharmacological pathway. The idea here is to make a drug that activates MOPs and produces all of the good and bad effects, but build into the drug a second activity that allows the good effects to proceed as normal but the bad effects to be suppressed. There are many variations of this kind of approach. Certainly, some analgesic drugs produce more than one effect. For example, the analgesic drugs tramadol and tapentadol both block the uptake of the neurotransmitter norepinephrine into nerve terminals in addition to activating MOR. Enhanced norepinephrine action in the spinal cord is thought to enhance the analgesic effects of MOR agonists, although not necessarily improve their profile.
But could a secondary action of a drug actually improve the profile of MOP agonists? This may be a possibility. For example, it has been reported that agonists that activate NOP receptors reduce the side effects of MOP agonists. So, why not make a drug that activates both of these receptors at the same time? This has been achieved and the initial results do appear to be very promising. The first NOP/MOP bifunctional drug of this type, named cebranopadol, appears to have good analgesic effects but few side effects, including lack of respiratory depression or abuse potential when tested in animal studies and, moreover, this desirable profile was also seen in human clinical trials to date. Such a drug would be a considerable advance if these results were confirmed. Encouragingly, a second NOP/MOP agonist named AT-121 has now been synthesized. The investigators tested the effects of AT-121 in monkeys using a tail withdrawal paradigm. The investigators placed monkeys’ tails in some uncomfortably warm water, which the monkeys normally only tolerated for a couple of seconds before removing their tails. However, after receiving AT-121 injections, monkeys kept their tails in the water for much longer periods of time, indicating that they didn’t feel the same degree of discomfort or pain. Even at higher doses, AT-121 did not cause the side effects that make most opioids so dangerous—suppression of breathing and heart rate, itch and physical dependence or tolerance. These results seem to be extremely impressive. So are NOP/MOP bifunctional agonists the answer to everybody’s opioid dreams? The results are certainly interesting. My problem with them is that there is no clear mechanism suggesting why NOP agonists should modify MOP effects in this way. There are numerous possibilities, of course, but the fact that there isn’t a clear answer makes me wonder. Nevertheless, the continued clinical progress of these drugs will be of considerable interest.
Another strategy that has been suggested relies on the relative distribution of MOPs in the nervous system. Many MOPs are situated in the central nervous system which is protected by what is known as the blood brain barrier (BBB). Many chemicals do not penetrate this barrier very easily, meaning that a chemical you eat or inject may not actually get into the brain, or will only do so with difficulty. However, some MOPs are also situated outside the BBB, on peripheral sensory nerves called nociceptors, whose job it is to send painful information into the spinal cord and the brain. How much of the analgesic effect of a drug like morphine depends on activating MOPs within the BBB and how much is due to activating the MOPs situated outside the BBB? If peripheral MOPs can produce significant amounts of analgesia, then one could potentially design a drug that only activates peripheral MOPs because it doesn’t pass through the BBB. As effects like drug abuse and respiratory depression are all mediated by MOPs within the central nervous system, a drug of this type should be able to produce a degree of analgesia with no central side effects. The only side effects it should produce would be things like constipation which result from the activation of MOPs outside of the brain. The potential success of this kind of selective targeting can be seen with the drug Targinact, which is a mixture of oxycodone and the opioid antagonist naloxone. Naloxone doesn’t get through the BBB very easily unless injected intravenously. Because of this, if it is given orally it will only block MOPs in the periphery and so will relieve the constipating effects of oxycodone which are produced by activating MOPs in the gut. There are animal studies which do suggest that activating peripherally located MOPs can produce some degree of analgesia. Moreover, MOP agonists already exist that don’t penetrate the BBB very easily. These are drugs like loperamide which are used as anti-diarrheal agents precisely because of their peripherally directed constipating effects. So, we might ask, do drugs like loperamide also produce analgesia? The answer is that they do appear to produce some degree of analgesia, and these effects may actually be more apparent under conditions of injury. Nevertheless, the degree of analgesia observed doesn’t seem to be in the same range as the profound effects of morphine. Some added benefit may be obtained by using peripherally directed MOP agonists that also activate other opioid receptors (KOPs and DOPs) or receptors for other agonists as well. For example, the drug DN-9, which is reported to be an effective and side-effect-free analgesic, activates peripheral MOPs, KOPs, DOPs and receptors for Neuropeptide F. However, human trials for such compounds are lacking.
It is clear that the goal of producing a version of morphine that is free of undesirable properties may be extremely challenging, and drugs for relieving pain may ultimately be better pursued on a disease-by-disease basis. For example, if we consider a painful disease like osteoarthritis, we know that the triggers for pain have some unique aspects, in this case degeneration of joint tissues, and this is also the case for other chronic pain syndromes. Thus, it may be that targeting the specific nociceptive triggers for different pain disorders will provide selective answers to ongoing pain issues. Of course, it may ultimately be possible to produce analgesic drugs that rival morphine and that are free from side effects like respiratory depression—but don’t hold your breath.