
The current opioid epidemic involves hundreds of thousands of individuals who have become addicted to opioids such as extended-release oxycontin. It should be remembered, however, that opioids are not just addictive recreational drugs. They also have an important bona fide use in medicine. Opioids are the most effective drugs we know of for the treatment of many kinds of pain, and normal surgeries—as practiced today in hospitals around the world—would be impossible without their use. Opioids are effective in controlling many types of pain. However, their chronic use almost inevitably leads to addiction. An important goal therefore is to produce drugs that effectively control long-term pain, but are free from the addictive potential of opioids. Since the advent of the opioid crisis, the National Institutes of Health (NIH), which is the main funding source for medical research in the United States, has been offering more research grants for people who are trying to develop “non-addictive”, but highly effective, analgesic drugs. One of the reasons why drugs like morphine are so good at controlling pain, and also why they have so many side effects, is that their receptors, known as μ-opioid receptors, are abundantly expressed at diverse points throughout the nervous system. These receptors are localized in peripheral nerves that transmit pain sensations, in the spinal cord, and in higher brain centers where they effectively control the flow of information along pain-conducting nerve pathways. Whether the receptors for other potential analgesics will be so conveniently placed is not at all clear, and so reproducing the powerful and widespread analgesic effects of opioids using other approaches may be difficult. Cannabis is one possibility, as cannabinoid receptors—like those for opioids—are widely expressed in the nervous system. Other approaches to the problem, such as synthesizing novel opioids that are free of addictive potential, have also proven to be challenging and, in spite of numerous claims in the scientific literature, have never ultimately succeeded. For reasons such as these, the replacement of opioids as generally effective analgesic drugs is a challenging undertaking.
On the other hand, all types of pain have unique triggers. For example, the trigger for osteoarthritic pain, involving movement of a degenerating joint, is different from that of painful diabetic neuropathy, involving dying peripheral neurons, or other causes. It is quite possible, therefore, that rather than discovering a replacement for opioids per se, new drugs for pain will be developed in a disease-specific manner, based on the particular pain-triggering mechanism that is involved in each syndrome. One example of such an approach is a new drug that is targeted to the treatment of sickle cell disease (SCD).
To understand this new therapeutic approach, we should first look at the mechanisms that underlie SCD, which has a particularly interesting origin. One of the greatest scourges known to man is the mosquito. The bite of the mosquito is associated with the transmission of a large number of diseases including dengue, West Nile virus, chikungunya, yellow fever, filariasis and zika, among others. However, by far the most prevalent and devastating of mosquito-associated diseases is malaria, which has tracked the development and migration of human society over millions of years. As described in a recent book (The Mosquito by Timothy C. Winegard), mosquitoes accompanied humans as they left Africa and colonized many other parts of the world. For example, the conquistadors brought malaria with them when they first came to the New World and it was diseases of this type, rather than their prowess as warriors, which were probably mostly responsible for their relatively easy conquest of Central and South America. Mosquitoes tend to breed in warm, marshy areas such as the Pontine Marshes surrounding the city of Rome which, in ancient times, were notorious for being a hot bed of malaria. Here again, it is quite possible that it was these malarial marshlands that protected Rome from many rampaging invading armies over the years.
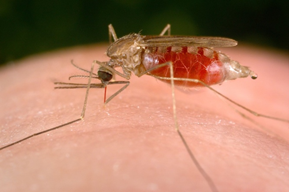
Prior to mosquitoes being identified as the “vector” that carried the disease, it was thought that malaria was spread by foul air—hence its name mal (bad)-aria (air). In actuality, malaria is spread by the bite of female Anopheles mosquitoes. The reason why female Anopheles mosquitoes bite you is to obtain your blood. Male and female Anopheles mosquitoes actually feed on nectar—blood isn’t what they eat. However, it turns out that there are nutrients in human blood—protein and iron—that are vital for the female Anopheles mosquito to be able to develop her eggs. This is the reason why she will drink about 3 millionths of a liter of human blood when she bites a human victim.
However, in addition to drinking your blood to obtain iron, mosquitoes also take up and inject disease-causing agents which basically go along for the ride. The infectious agent that causes malaria is a protozoan (single-celled eukaryotic organism), and we have a detailed description of its entire life cycle. There are actually several closely related protozoan parasites that can cause the disease. Moreover, there are also different species of mosquitoes that can spread malaria. However, only different types of female Anopheles mosquitoes can transmit human malaria. The organisms involved are 5 species of the protozoan Plasmodium—P. vivax, P. falciparum and P. malariae being the most common forms of the disease in humans. Related protozoa can infect other animals including birds and even fish. When a female mosquito bites somebody, if her victim is infected with a malaria parasite—let us assume it is P. falciparum as an example—then she will also take up male and female protozoan gametocytes (precursors of sperm and eggs) that circulate in the victim’s peripheral blood. Once within the mosquito, these gametocytes will develop further into male and female gametes (the sperm and eggs). The motile male gamete seeks out a female gamete and fertilization results in a motile zygote, also known as an “ookinete,” formed within the lumen of the mosquito’s gut. The ookinete then penetrates the gut wall where it becomes an “oocyst.” This then multiplies, forming infectious “sporozoites” that migrate to the salivary glands of a mosquito and are injected when the mosquito bites to obtain her next blood meal. Once the infectious sporozoites have entered the blood of the victim, they circulate around the body, eventually taking up residence in liver cells where they undergo further multiplication (exoerythrocytic schizogony), producing the next stage of development: the merozoites. Merozoites exit the liver into the blood and they are the species which now infect red blood cells (erythrocytes). Here, they multiply further to produce more merozoites which then destroy the erythrocyte and enter the blood once more and infect further erythrocytes. This process continues ad infinitum, until the host’s blood is used up and the individual dies. During the time when merozoites are multiplying within red blood cells, some of them turn into gametocytes that can be taken up by a mosquito during a blood meal to be transferred to the next victim. And so it goes. Around and around in a horrible dance of death and destruction reminiscent of the most terrifying scenes of the classic horror movie, Alien.
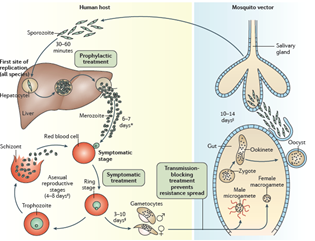
The well-known bouts of fever associated with human malaria result from rounds of bursting of red blood cells and release of infectious particles and toxins. The different types of Plasmodium parasites have slightly different properties in this regard generating “tertian” or “quartan” fevers which occur on average every two or three days. P. falciparum is the most deadly form of the disease. Some types of malaria such as P. vivax can also produce forms that lie dormant in the liver (hypnozoites) even when the disease has been cleared, only to awaken again after many weeks to initiate another deadly round of fevers, vomiting, and other symptoms of the disease.
The powerful selection pressures on individuals living with the constant threat of malaria in Sub-Saharan Africa led to the appearance of a genetic trait some 7,300 years ago that afforded some resistance to P. falciparum. This trait was caused by a mutation in the protein hemoglobin, the major protein found in red blood cells whose job is to carry oxygen from the lungs to the rest of the body. The most common mutation is known as sickle hemoglobin (HbS). HbS is a structural variant of normal adult hemoglobin. Adult hemoglobin (HbAA) is made up of two α and two β globin chains. HbS is the result of a single point mutation (Glu → Val) on the sixth codon of the β-globin gene. People who are heterozygous for the HbS allele carry the sickle cell trait (HbAS) but do not have SCD, whereas individuals who are homozygous for the HbS allele have SCD of which sickle cell anemia (SCA) is the most common manifestation. SCA is characterized by chronic hemolytic anemia, unpredictable episodes of pain, and widespread organ damage.
SCD is one of the most common genetic disorders found in the world. An estimated 20–25 million people live with homozygous SCD (HbSS). Approximately 300,000 infants are born annually with HbSS. Areas with a high prevalence of malaria such as Sub-Saharan Africa, the Mediterranean basin, Middle East, and India tend to have higher populations of patients affected with SCD. In the United States, approximately 100,000 people—many of them the descendants of the African slave trade—have SCD, accounting for more than 110,000 sickle cell-related hospitalizations annually. There is a wide variability in the clinical severity of SCA, as well as in the life expectancy. Interestingly, although people who carry the HbAS trait (heterozygotes) are clearly resistant to malaria, the mechanism of this effect isn’t entirely clear. Different kinds of biochemical changes have been detected in red blood cells from HbAS individuals, which must make it harder for P. falciparum to establish an infection but there is no real consensus as to how this occurs. Clearly, SCD is an extremely serious health problem. The fact that it has been maintained in populations such as those in Sub-Saharan Africa where malaria is endemic, speaks to the fact that just about anything is better than having malaria.
Individuals who are homozygous for hemoglobin S (HbSS), having two affected β-chains, develop SCD. The HbS allele has a lower-than-normal affinity for oxygen and also has a tendency to aggregate at low oxygen concentrations. Hence, when red blood cells have delivered their oxygen to the tissues and are returning to the lungs via the venous system in an oxygen-depleted state, the hemoglobin of SCD patients polymerizes, which produces sickle-shaped malfunctioning erythrocytes and reticulocytes (erythrocyte precursors). These cells are sticky and form clumps that occlude blood vessels, particularly small and some large vessels producing what is known as a vaso-occlusive crisis (VOC) which causes ischemic injuries. The sickled red blood cells are also brittle and burst, producing profound hemolytic anemia. During a VOC, the most common complaint is pain, and recurrent episodes may cause irreversible organ damage. One of the most severe forms is the acute chest syndrome which occurs as a result of infarction of the lung parenchyma. This can rapidly result in death. For a patient with SCD, both chronic pain and intense bouts of acute pain are particularly hard to deal with. Traditionally, patients with SCD-associated pain are treated with opioids and around a quarter of patients use opioids chronically throughout life, but of course, this also comes with the well-known risks of chronic opioid use.
In order to produce an alternative to opioid use, we need to know exactly why pain occurs in SCD. Nerves that sense pain innervate blood vessels and can be activated by inflammatory mediators and other processes occurring during a VOC. Drugs that interfere with these processes may be helpful in treating pain occurring in SCD patients in particular. Recently, there has been progress in achieving this aim. The first new therapy to be introduced for treating SCD was hydroxyurea (or hydroxycarbamide), which is the only drug frequently employed in SCD management that is approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Hydroxyurea’s principal mechanism of action is stimulating the production of HbF: a fetal form of Hb. When HbF is produced, it reduces HbS polymerization and so ameliorates pathological processes such as sickling, VOCs, and events downstream of these triggers. The mechanism by which hydroxyurea induces HbF generation is unclear. Since 1998, hydroxyurea has been the only FDA-approved therapy for SCD. By increasing both fetal hemoglobin concentrations in erythrocytes and reticulocytes, hydroxyurea treatment has significantly reduced the rates of VOCs, subsequent hospitalizations, and mortality in a range of patients. Unfortunately, despite these benefits, adherence to a hydroxyurea regimen has been problematic for some patients. Adverse effects of the drug include bone marrow suppression, large inter-patient variability in the effects it produces, and variations in the maximum tolerable dose. Consequently, other therapies have been sought based on a detailed understanding of the molecular and cellular events that produce VOCs.
Disturbances in blood flow produced by phenomena such as red cell sickling can be “sensed” by the endothelial cells that line blood vessels. Circulating sickled blood cells adhere to each other and to the activated endothelium, contributing to and potentially initiating VOCs. Activation of endothelial cells involves a large number of cellular responses that have pro-inflammatory consequences. For example, in postcapillary venules, activated endothelial cells express the proteins P‑selectin and E‑selectin. These are lectin-like molecules that can bind to specific patterns of sugar residues attached to proteins on the surfaces of circulating cells, allowing them to leave blood vessels and enter tissues as part of the inflammatory response. Activated platelets and adhesive sickle erythrocytes can adhere to circulating or endothelium-bound cells like neutrophils and form aggregates. Sickle erythrocytes can also bind directly to the activated endothelium. Endothelial cell activation—which occurs as a consequence of endothelium-derived nitric oxide (NO) depletion by cell-free hemoglobin released during hemolysis—oxidative stress, and constant inflammatory processes all play a key role in the initiation of VOCs. The adhesion of red blood cells and leukocytes to the activated endothelium acts as a trigger for VOCs.
These mechanisms means that P-selectin may well be an important drug target in situations like SCD. Over the past 25 years, selectins—particularly P-selectin—have been shown to contribute to pathological inflammation and thrombosis in many preclinical disease models, including atherosclerosis, ischemia–reperfusion injury, arterial thrombosis, and deep vein thrombosis. Patients with SCA develop injury to many organs that is consistent with repeated episodes of ischemia–reperfusion injury caused by the occlusion of blood vessels in VOC. Models of sickle cell anemia in experimental animals have established that sickled red cells obstructing small vessels trigger pathological inflammation and thrombosis that involves adhesive interactions among red cells, platelets, and the endothelium. P-selectin on the surface of activated platelets and endothelial cells binds directly to ligands on sickled red cells and immune cells like neutrophils. Blockade of P- or E-selectin decreases these adhesive interactions and improves microcirculatory flow in sickle cell mice. Results such as these suggest that agents that interfere with the functions of P-selectin might have value in preventing VOCs in SCD, and such an approach has recently been shown to have considerable promise.
Crizanlizumab is a recently developed humanized monoclonal antibody (a “mab”) that binds to P-selectin on the surface of endothelial cells and platelets and blocks its interaction with other cells. A large international clinical trial investigated the effects of crizanlizumab therapy on the rate of VOCs in a total of 198 participating SCD patients (aged 16–56 years), during 52 weeks of treatment. The intravenous administration of the P-selectin inhibitor was well tolerated, and high-dose crizanlizumab (5 mg/kg) was associated with a significantly lower frequency of VOCs in the treated patients, compared with the placebo group, where the median rate of VOCs per year was 1.63 versus 2.98 with placebo. Additionally, the median time to first crisis was significantly longer with 5 mg/kg crizanlizumab than with placebo (4.07 vs. 1.38 months). With VOCs being the major contributing factor to hospitalizations and reduced patient quality of life, these results were considered to be extremely promising and the United States Food and Drug Administration granted crizanlizumab a “Breakthrough Therapy” designation for the prevention of VOCs in SCD, implying that crizanlizumab demonstrated substantial improvement over existing therapies based on the available preliminary clinical evidence. On November 15, 2019, the FDA approved crizanlizumab-tmca (ADAKVEO, Novartis) for use in the reduction of the frequency of VOCs in adults and pediatric patients aged 16 years and older with sickle cell disease.
The FDA approval of crizanlizumab is hopefully good news for SCD sufferers. It also illustrates the important principle of treating pain on a disease-by-disease basis rather than going for a “home run” knockout drug that will treat more or less everything. This is what happened in the field of cancer. In the end, there was no “cure for cancer.” What we have achieved is a piecemeal approach where different cancers are targeted with different types of therapeutic agents depending on their particular mechanisms. There are plenty of painful diseases that might be helped by such an approach. Consider Ehlers-Danlos syndrome (EDS), for example. Like SCD, EDS is a genetically inherited disease which, in this instance, attacks connective tissue rather than the blood. Like those with SCD, EDS patients are afflicted with chronic pain that is often treated by long term use of opioids. However, judging by the success of crizanlizumab for SCD, we can easily imagine producing a drug for EDS pain that was directly targeted to a specific trigger. Moreover, there are many other painful diseases of this type. It is unlikely that we will ever improve on opioids for some types of pain but, as for many other situations, progress may well be made one step at a time.